Linux NFS服務搭建與實戰:構建高效數據處理與存儲服務
一、引言
在數據處理和存儲服務架構中,實現多服務器間的文件共享是提高工作效率、降低存儲成本的關鍵。Network File System(NFS)作為一種成熟、高效的分布式文件系統協議,允許網絡中的客戶端計算機像訪問本地存儲一樣訪問遠程服務器上的文件系統。本文將詳細介紹如何在Linux環境下搭建NFS服務,并展示其在數據處理和存儲服務中的實際應用案例。
二、NFS服務原理與優勢
1. NFS工作原理
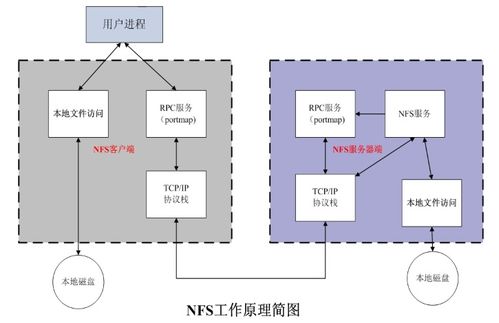
NFS基于客戶端-服務器架構,服務器端將特定的目錄共享(導出)給網絡中的授權客戶端。客戶端通過掛載這些遠程目錄到本地文件系統,實現透明訪問。NFS默認使用TCP/UDP的2049端口,依賴于RPC(遠程過程調用)機制進行通信。
2. NFS在數據處理與存儲中的優勢
- 集中化管理:數據統一存儲在NFS服務器,便于備份、維護和權限控制
- 跨平臺兼容:支持多種操作系統訪問同一數據源
- 高性能共享:多個計算節點可同時訪問同一數據集,適合分布式計算場景
- 存儲成本優化:避免數據在多節點間的冗余存儲
- 簡化部署:應用程序和配置文件可集中存放,各節點無需單獨部署
三、NFS服務搭建步驟
1. 環境準備與軟件安裝
`bash
# 在NFS服務器端安裝必要軟件包
CentOS/RHEL系列
yum install nfs-utils rpcbind -y
Ubuntu/Debian系列
apt-get install nfs-kernel-server -y`
2. 配置NFS服務器
`bash
# 創建共享目錄
mkdir -p /data/nfsshare
chmod 777 /data/nfsshare # 根據實際需求設置權限
配置導出目錄(/etc/exports)
格式:共享目錄 客戶端IP(選項)
示例:允許192.168.1.0/24網段讀寫訪問
/data/nfsshare 192.168.1.0/24(rw,sync,noroot_squash)
常用選項說明:
rw:讀寫權限
ro:只讀權限
sync:同步寫入,數據安全但性能較低
async:異步寫入,性能高但風險較大
norootsquash:信任客戶端root用戶
all_squash:將所有用戶映射為匿名用戶
`
3. 啟動與驗證服務
`bash
# 啟動相關服務
systemctl start rpcbind
systemctl start nfs-server # 或nfs-kernel-server(Ubuntu)
systemctl enable rpcbind nfs-server
驗證服務狀態
systemctl status nfs-server
rpcinfo -p localhost
查看當前導出列表
exportfs -v
showmount -e localhost`
4. 客戶端配置
`bash
# 安裝客戶端軟件
CentOS/RHEL
yum install nfs-utils -y
Ubuntu/Debian
apt-get install nfs-common -y
創建本地掛載點
mkdir -p /mnt/nfs_client
掛載NFS共享目錄
mount -t nfs 服務器IP:/data/nfsshare /mnt/nfsclient
驗證掛載
df -hT | grep nfs
mount | grep nfs
設置開機自動掛載(/etc/fstab)
服務器IP:/data/nfsshare /mnt/nfsclient nfs defaults 0 0`
四、數據處理與存儲服務應用案例
案例1:分布式計算數據共享平臺
場景:生物信息學分析中,多個計算節點需要訪問同一基因組數據庫
解決方案:`bash
# NFS服務器配置
/etc/exports 添加以下配置
/share/genomedb 192.168.10.0/24(ro,sync,allsquash) # 只讀共享參考基因組
/share/rawdata 192.168.10.0/24(rw,sync,norootsquash) # 讀寫共享原始數據
/share/results 192.168.10.0/24(rw,sync,noroot_squash) # 結果輸出目錄
客戶端使用示例
在各計算節點掛載共享目錄
mount -t nfs nfs-server:/share/genomedb /reference
mount -t nfs nfs-server:/share/rawdata /data/input
mount -t nfs nfs-server:/share/results /data/output
運行分析流程(示例)
bwa mem /reference/hg38.fasta /data/input/sample1.fastq > /data/output/sample1.sam`
效益:
- 參考基因組只需存儲一份,節省80%存儲空間
- 分析結果集中存儲,便于統一管理和后續處理
- 新計算節點加入無需數據遷移,快速擴展計算能力
案例2:企業級日志收集與分析系統
場景:多臺Web服務器日志需要集中存儲并進行實時分析
解決方案:`bash
# NFS服務器配置高可用方案
使用DRBD+Heartbeat實現NFS高可用
/etc/exports 配置
/var/log/centralized 192.168.20.0/24(rw,sync,nosubtreecheck,norootsquash)
Web服務器客戶端配置
配置rsyslog將日志實時寫入NFS
/etc/rsyslog.conf 添加
. @nfs-server:/var/log/centralized
或使用logrotate直接寫入
/var/log/httpd/*.log {
sharedscripts
postrotate
cp /var/log/httpd/access.log /mnt/nfslogs/webserver-$(hostname)-access.log
cp /var/log/httpd/error.log /mnt/nfslogs/webserver-$(hostname)-error.log
endscript
}`
效益:
- 實現日志集中管理,便于安全審計
- 支持實時日志分析工具(如ELK Stack)直接處理
- 避免單點故障,確保日志服務高可用性
案例3:機器學習訓練數據共享
場景:多GPU服務器需要訪問大型訓練數據集
解決方案:`bash
# 針對大文件優化NFS配置
/etc/exports 使用異步寫入提高性能
/share/datasets 192.168.30.0/24(rw,async,nowdelay,noroot_squash,insecure)
調整內核參數優化性能
/etc/sysctl.conf 添加
增加NFS讀寫緩沖區大小
sunrpc.tcpslottableentries = 128
sunrpc.udpslottableentries = 128
客戶端掛載優化選項
mount -t nfs -o rsize=32768,wsize=32768,timeo=600,nolock \
nfs-server:/share/datasets /datasets
分布式訓練示例(PyTorch)
import torch
from torch.utils.data import DataLoader
dataset = CustomDataset('/datasets/imagenet/')
dataloader = DataLoader(dataset, batchsize=256, numworkers=8)`
效益:
- 多個訓練節點可同時訪問數百GB的圖像數據集
- 避免數據復制,節省SSD存儲壽命和網絡帶寬
- 支持大規模分布式訓練架構
五、性能優化與安全建議
1. 性能調優
`bash
# 服務器端優化
增加NFS線程數(/etc/sysconfig/nfs)
RPCNFSDCOUNT=32
客戶端優化
調整掛載參數
mount -t nfs -o \
rsize=65536,wsize=65536,hard,intr,timeo=600,retrans=2 \
服務器IP:/共享目錄 /本地目錄`
2. 安全加固
`bash
# 使用防火墻限制訪問
iptables -A INPUT -s 允許的IP段 -p tcp --dport 2049 -j ACCEPT
iptables -A INPUT -p tcp --dport 2049 -j DROP
配置更精細的訪問控制
/etc/exports 示例
/share/sensitive 192.168.1.100(rw,sync) 192.168.1.101(ro,sync)
啟用NFSv4,支持Kerberos認證
服務器配置(/etc/sysconfig/nfs)
RPCNFSDARGS="-V 4.2"
客戶端掛載
mount -t nfs4 -o sec=krb5 服務器IP:/共享目錄 /本地目錄`
3. 監控與維護
`bash
# 監控NFS性能
nfsstat -c # 客戶端統計
nfsstat -s # 服務器端統計
查看連接狀態
netstat -an | grep 2049
日志檢查
tail -f /var/log/messages | grep nfs`
六、常見問題排查
- 掛載失敗:訪問被拒絕
- 檢查/etc/exports配置是否正確
- 確認防火墻未阻止2049端口
- 驗證showmount -e 服務器IP 能否看到共享
- 讀寫性能差
- 調整rsize/wsize參數
- 考慮使用async選項(注意數據一致性風險)
- 檢查網絡帶寬和延遲
- 客戶端卡住或無響應
- 使用hard掛載選項(默認)配合timeo參數
- 檢查服務器負載和存儲性能
- 考慮實施NFS高可用方案
七、
NFS作為成熟的網絡文件系統解決方案,在數據處理和存儲服務中發揮著重要作用。通過合理配置和優化,NFS能夠為分布式計算、日志管理、機器學習等場景提供高效、可靠的文件共享服務。在實際部署中,需要根據具體應用場景平衡性能、安全性和可用性要求,并建立完善的監控和維護機制,確保服務穩定運行。
隨著技術的發展,用戶也可以考慮將NFS與更現代的分布式文件系統(如Ceph、GlusterFS)結合使用,或直接遷移到云原生存儲方案,以滿足更大規模、更復雜的數據處理需求。